
Can Data Just Speak for Itself?
Gosh did I find a surprise looking at the original carvedilol trials in heart failure.

Tomorrow over at CardiologyTrials we will describe the CAPRICORN trial of carvedilol vs placebo in patients post myocardial infarction. Carvedilol is now widely accepted for this indication, but CAPRICORN featured a super-interesting twist regarding how we interpret data given pre-experiment choices. Do tune in. (Also, the bad typo in the original e-mail is fixed.)
This discovery led me to an even more interesting twist regarding the first Carvedilol trial in heart failure. Here is a screenshot of the trial from NEJM in 1996.

I want to state clearly now that I do not know the right resolution to the main question of this post. I am not sure there is a right answer.
The main conclusion of this seminal paper was that carvedilol reduces death and the risk of hospitalization for cardiac reasons.
The trial enrolled about a 1100 patients with 2:1 randomization; about 700 in the carvedilol arm and 400 in the placebo arm. “There were 31 deaths (7.8 percent) in the placebo group and 22 deaths (3.2 percent) in the carvedilol group.” This represented a 65% decrease in death and the 95% confidence intervals went from a 39% to an 80% decrease in death. The p-value calculated at less than 0.001.
It sounds like a no-brainer, doesn’t it? Yet the first time it went to an FDA advisory committee meeting it was voted down—definitively.
How could that be?
Well, it turns out that there were 4 US carvedilol trials in HF. Three of the four, including this one, had a functional endpoint of exercise tolerance. None of these three trials met statistical significance for this endpoint. Carvedilol did not help patients with heart failure feel better.
But there is even more. In the above seminal trial, total mortality was not selected as a primary or secondary endpoint. Experts in HF, therefore, at the time, must not have expected it.
The difference in total mortality was a surprise finding.
The first FDA advisory committee had many statisticians on it. Their thinking, summed up by an amazingly clear piece by Dr. Lemuel Moye, goes something like this:

Scientists must declare a primary endpoint before the experiment. This, Moye writes, is the access which a trial revolves around.
That is not what happened in the case of the US carvedilol trials. The investigators, and to be honest me as well, wondered why you would not just let the data speak for itself. Such an important death signal cannot be ignored.
I wrote about this issue to regulatory expert Dr. Sanjay Kaul, who sent me two examples wherein an after-the-fact surprise mortality signal was NOT confirmed.
One was vesnarinone and the other amlodipine.

You can see from this half slide that both drugs in their first trial found huge reductions in death. In both cases however, total mortality was not the primary endpoint.
Here is the slide with the larger studies. You can see that neither signal panned out in subsequent larger studies.
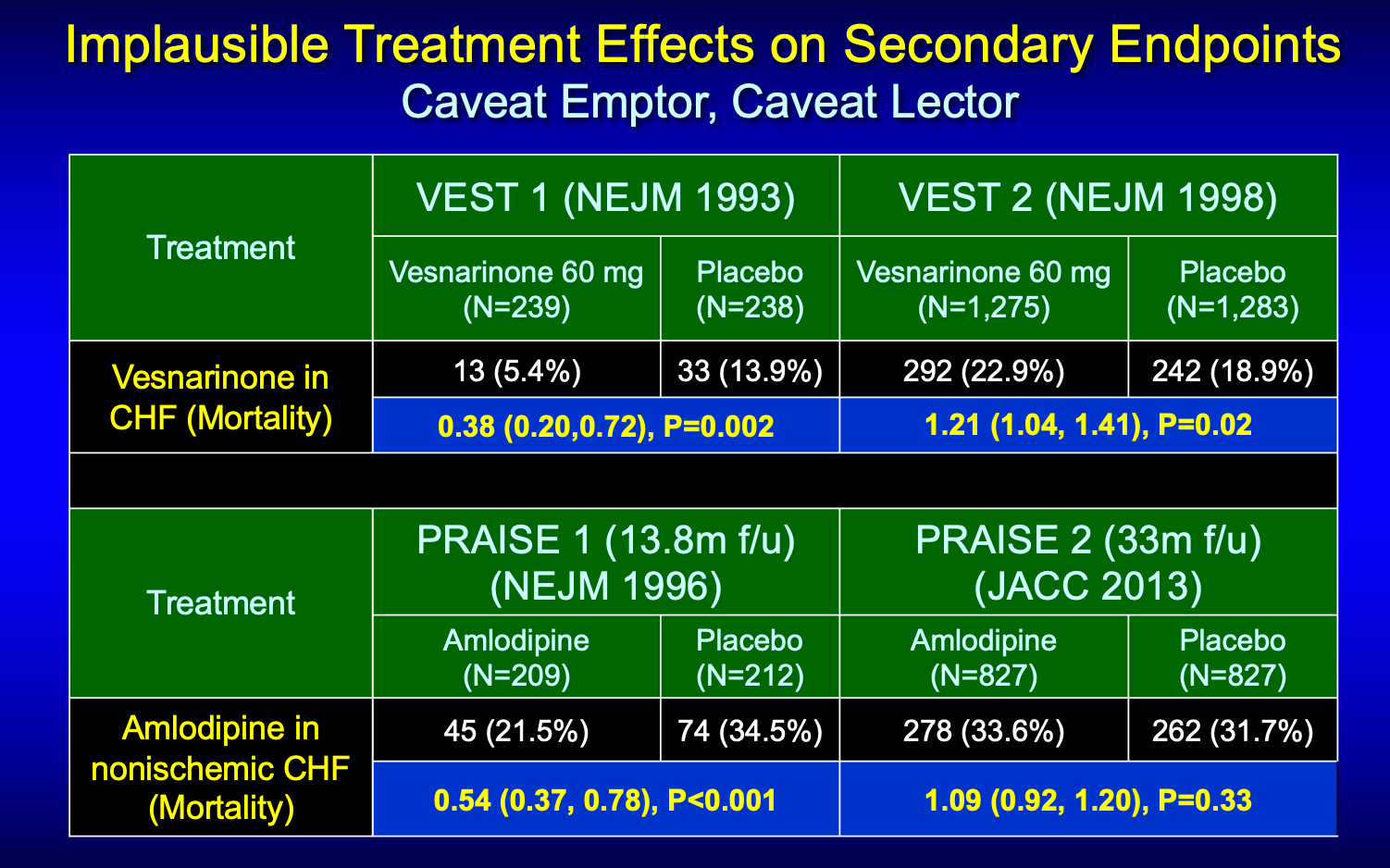
In the carvedilol case, the investigators asked us to ignore the negative primary endpoint of the trials (exercise tolerance) and focus on the mortality finding. Because it is a) important, b) a big decrease, and c) statistically persuasive.
Dr Moye disagreed. He likens ignoring the negative primary endpoint as the “crazy aunt in the attic.” He explained that an experiment has a glassful of false positive chances. If you use part of it up in the primary endpoint, there is only so much left to use in subsequent endpoints.
In the carvedilol example the p-value for the primary endpoint was over 0.05 so all the false positive chances were completely used. And, thus, the downstream mortality finding was highly likely to be a false positive—akin to the vesnarinone and amlodipine example I just showed you.
Moye worried greatly about foisting false findings into the public realm.
In the end, Moye’s worries were not realized. A subsequent carvedilol study proved the drug reduced mortality in heart failure. Two other beta-blockers, metoprolol succinate and bisoprolol were also shown effective in reducing death in stable outpatients with heart failure.
The Take-Home Lessons
I’ve been showing the beta-blocker heart failure trials as examples of clear signals of benefit. And they ended up being so.
But I did not know that the first trials were actually negative for their primary endpoint. Investigators surprisingly noted the lower signal of mortality. They let the data speak for itself.
Carvedilol in stable outpatients with heart failure turned out not to be vesnarinone or amlodipine, but this was not known at the time of the second FDA meeting in 1997 when a more clinician-heavy committee approved carvedilol.
When we show CAPRICORN tomorrow on CardiologyTrials, a similar story will come up. That is, what should we think of surprising results downstream from a nonsignificant primary endpoint?
I don’t have the answer. Some of it involves philosophy. Namely, what is worse, letting an uncertain potentially harmful therapy out in society, or holding back a potentially beneficial one?
For me, I lean on the first do no harm principle. Proponents of a new therapy ought to show us their intervention works in a proper way—via the primary endpoint.
The second question I pose pertains to use of statistical principles.
A reader of that first carvedilol trial in NEJM in 1996 would have little idea that the authors made quite a contentious statistical leap. They were proven correct, but this was not known at the time.
I think that there were many papers about this case in Statistics in Medicine. This case is all about the illogic of multiplicity adjustments when one wants to ask specific questions about specific endpoints. Other than in frequentist statistics, rules of evidence are much clearer than this discussion would have you believe. For example, if the police decide that the first suspect in a crime is not the best suspect and they arrest a second person, there is no logic in downweighting the fingerprint and motives of the 2nd suspect just because there was an earlier suspect. Evidence of guild for the 2nd suspect must come from evidence about THAT suspect and the detective’s prior beliefs. The logical Bayesian approach would be to formulate prior distributions of effects of the drug, separately for each endpoint, before data are available. Then apply those priors to the data and don’t look back. Even better: create a hierarchical ordinal endpoint for judging which group of patients fared better overall. Death would be at the top of the scale, and the ordinal analysis would penalize or reward for death even though the sample size may have been inadequate for judging mortality on its own.
Let's get philosphical Dr. Mandrola.
Here is a study:
standard of care plus placebo "b" (blue and smaller pill)
vs.
standard of care plus placebo "R" (red and bigger pill)
Placebo "b" 7.8% deaths (out of 400)
Placebo "R" 3.1% deaths (out of 700)
Is it a "no brainer" to start using Placebo "R"? Why? I suppose that depends on what we mean by "no brainer".
Placebo "b" costs $0.01, Placebo "R" costs $20.00. Now what?